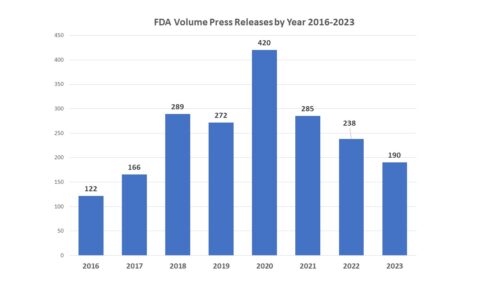
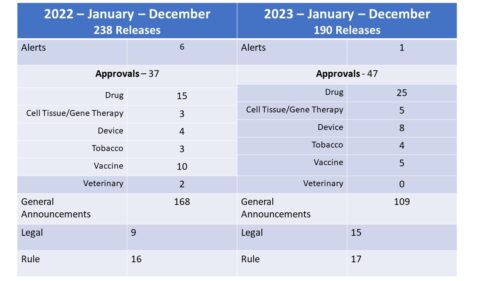
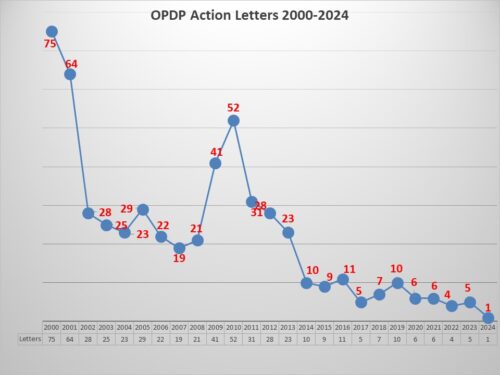
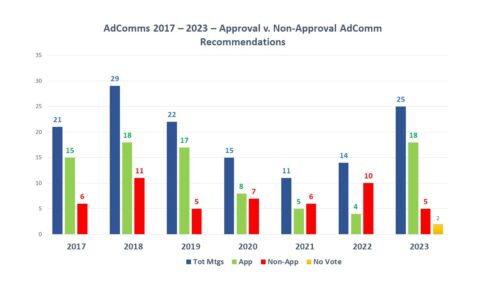
Back in December 2023, FDA announced intention in the Federal Register and in a press release to form a new FDA Advisory Committee to be called the Genetic Metabolic Diseases Advisory Committee (GeMDAC). As noted in a recent posting here, last year saw a marked increase not only in the number of advisory committee meetings, but in the proportion of recommendations for approval.
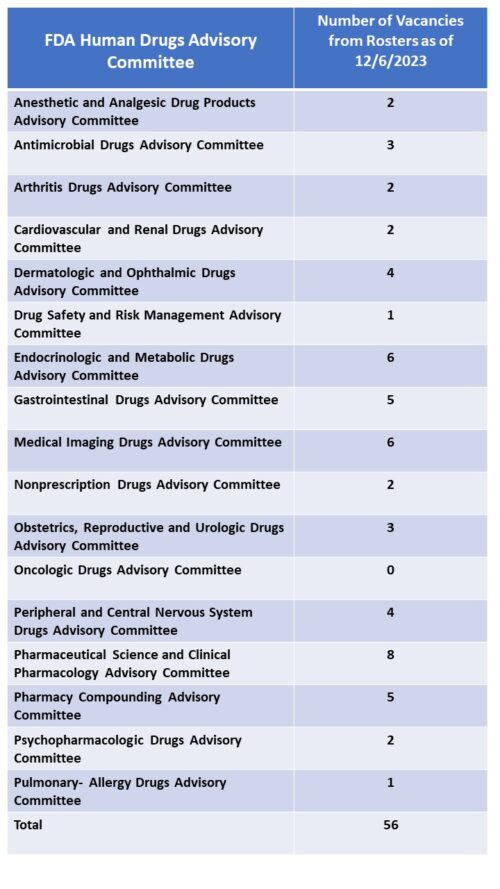
FDA currently has over 30 advisory committees providing the agency with access to advice from experts outside of government to help in evaluating new therapies for safety and efficacy as well as defining policy positions around many of the principles that guide such decisions. There are 18 committees that are focused on human drugs – organized by subject matter jurisdiction – such as the Arthritis Drugs Advisory Committee, the Cardiovascular Drugs Advisory Committee along with several others. These committees are called upon by FDA to frequently (though not always) provide input into the approval consideration for new drugs which often (again not always) culminate in a defining vote among the members to support conclusions related to safety and efficacy. The vote of any committee is advisory in nature, and not binding on the agency’s final decision.
Per the Mayo Clinic, there are a host of metabolic disorders – conditions caused by an inherited genetic disorder that impact the way we break down food to make energy or help us rid ourselves of substances we no longer need in our bodies – and include conditions such as Gaucher Disease, Niemann-Pick, Tay-Sachs disease, and Wilson’s disease.
The new committee falls under the purview of the of the Division of Rare Diseases and Medical Genetics which was established in 2020 within the Center for Drug Evaluation and Research (CDER) Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine (ORPURM) in 2020. According to FDA the myriad of different genetic metabolic diseases are often rare diseases that can have both high morbidity and negatively impact quality of life.
Since 1983 with the passage of the Orphan Drug, there has been increased attention and focus to bringing new treatments into play for rare diseases, with increasing numbers of new orphan drug approvals seen between 2013-2022 according to the National Institutes of Health. And also according to NIH, drugs addressing metabolic disorders were among the fourth highest number of new orphan drug approvals between 1983 and 2022 (see Table 1), following orphan drugs in oncology, neurology and infectious diseases. The development of an advisory committee with this jurisdiction might indicate an expectation that the pipeline in this space may require FDA to convent advisory committees to consider increasing numbers of new approvals, perhaps with increased complexity associated with them.
GeMDAC at this time does not have any members appointed. According to the agency the new members will be announced on the FDA’s website where the roster is housed “once all members have been appointed” and a specific date for accomplishing this is not known, however in response to an email inquiry, the agency said that the expectation is that the committee will be ready to meet by the time that a particular matter is in need of the committee’s input. Anyone interested in submitting a nomination for consideration for the committee, which will have 9 members and will include a member from industry and a consumer representative, can do so by accessing the link in the agency press release about the committee establishment or by visiting the GeMDAC web page. Another advisory committee established late in 2023 – the Digital health Advisory Committee – is also looking for members.